15号染色体异常
人类通常在每个细胞中有46条染色体,分为23对。15号染色体的两个拷贝,一个从每个亲本遗传的拷贝,形成一对。15号染色体跨越超过1.02亿个DNA构建块(碱基对),占细胞总DNA的3%以上。
人类通常在每个细胞中有46条染色体,分为23对。15号染色体的两个拷贝,一个从每个亲本遗传的拷贝,形成一对。15号染色体跨越超过1.02亿个DNA构建块(碱基对),占细胞总DNA的3%以上。
识别每条染色体上的基因是遗传研究的一个活跃领域。由于研究人员使用不同的方法来预测每条染色体上的基因数量,因此估计的基因数量会有所不同。15号染色体可能包含600至700个基因,这些基因提供制造蛋白质的说明。这些蛋白质在体内发挥着各种不同的作用。
15号染色体位图
遗传学家使用称为idiograms的图表作为染色体的标准表示。图像显示染色体的相对大小及其条带图案,这是当染色体用化学溶液染色然后在显微镜下观察时出现的暗带和亮带的特征图案。这些条带用于描述每条染色体上基因的位置。
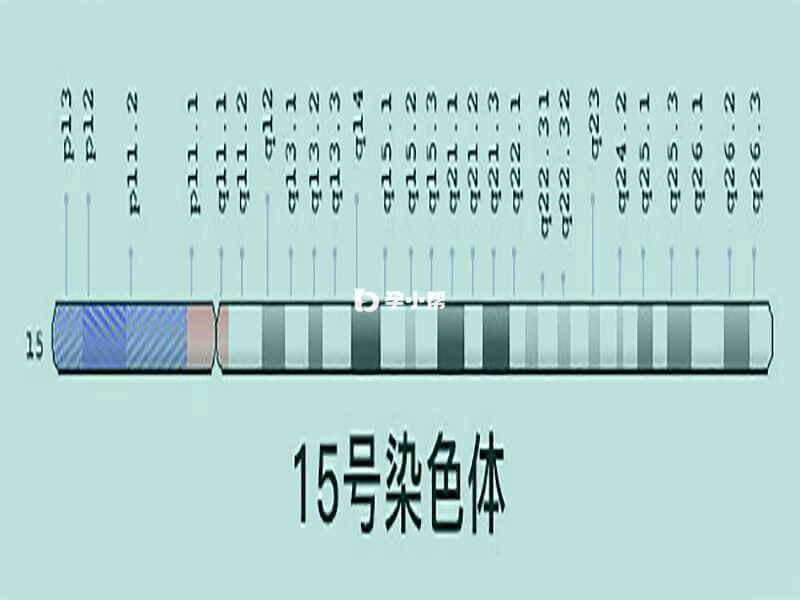
15号染色体结构的其他变化可以具有多种效果,包括智力残疾,发育迟缓,独特的面部特征,出生缺陷和其他健康问题。15号染色体的变化包括每个细胞中染色体的p臂或q臂的额外(重复)或缺失区段。额外或删除的细分的大小各不相同;所涉及的遗传物质的数量有助于产生的体征和症状。
15号染色体异常的相关疾病
15号染色体是承载DNA非常重要的一个载体,15号染色体跨越超过1.02亿个DNA构建块(碱基对),占细胞总DNA的3%以上。如果15号染色体发生异常则会导致一系列病症,如发育迟缓、智力残疾以及行为或精神疾病等。具体如下:
15q13.3微缺失症
15q13.3微缺失是染色体变化,其中在每个细胞中缺失一小块15号染色体。缺失发生在染色体的长(q)臂上指定为q13.3的位置。大多数具有15q13.3微缺失的人缺少大约200万个DNA构建块(碱基对)的序列,也写成2兆碱基(Mb)。删除区域的确切大小各不相同,但通常包含至少6个基因。

其他15q13.3微缺失的患者没有明显的与染色体变化相关的体征或症状。在这些个体中,微缺失通常在进行基因检测时被检测到,因为它们具有受影响的亲属。目前尚不清楚为什么15q13.3微缺失导致某些人的认知和行为问题,而其他人的健康问题很少或没有。研究人员认为可能涉及其他遗传或环境因素。
15q24微缺失症
15q24微缺失是染色体变化,其中每个细胞中缺失一小块15号染色体。具体而言,受影响的个体在15号染色体上q24位的DNA缺失1.7Mb和6.1Mb之间。缺失的确切大小不同,但所有个体缺失相同的1.2Mb区域。该区域包含几个被认为对正常发育很重要的基因。
急性早幼粒细胞白血病
一种称为急性早幼粒细胞白血病的血癌是由15号染色体和17之间遗传物质的重排(易位)引起的。这种易位,写为t(15;17),融合了部分15号染色体的PML基因。来自染色体17的RARA基因的这种突变是在一个人的一生中获得的,并且仅存在于某些细胞中。
这种称为体细胞突变的遗传变异不是遗传的。t(15;17)易位被称为平衡的相互易位,因为染色体彼此交换(互逆)并且没有遗传物质被获得或丢失(平衡)。由该融合基因产生的蛋白质称为PML-RARα。
等二尖瓣15号染色体综合征
等二尖瓣15号染色体综合征是由每个细胞中存在一个异常的额外染色体(称为阳极中心15号染色体)引起的。等距中心染色体包含遗传物质的镜像区段,并且具有两个收缩点(着丝粒),而不是正常染色体中的一个着丝点。
Prader-Willi综合征
Prader-Willi综合征是由15号染色体区域的活性基因缺失引起的。该区域位于染色体的长(q)臂上,命名为15q11-q13。虽然不同的基因与这两种疾病有关,但是15号染色体的同一部分通常会受到Angelman综合征患者的影响。

人们可能患有Prader-Willi综合征或Angelman综合征,但他们通常不能兼得。人们通常从每个父母那里继承一份15号染色体。这条染色体上的一些基因仅在遗传自人父亲(父本)的拷贝上开启(活跃)。这种亲本特异性基因激活是由称为基因组印记的现象引起的。
感觉神经性耳聋和男性不育
感觉神经性耳聋和男性不育是由于15号染色体[1]的长(q)臂遗传物质的缺失引起的。感觉神经性耳聋和男性不育症的症状与该地区多个基因的丧失有关。删除的大小因受影响的个体而异。研究人员已经确定,15号染色体STRC上特定基因的丢失是受影响个体听力损失的原因。

在15号染色体的同一区域中另一个基因CATSPER2的丢失是造成精子异常的原因,这导致受影响的男性无法生育孩子(不育)。研究人员正在努力确定缺失区域中其他基因的丢失如何影响患有感音神经性耳聋和男性不育的人。
15号染色体数量或结构的其他变化可导致智力残疾,延迟生长发育,肌张力减退和面部特征。这些变化包括每个细胞中部分15号染色体的额外拷贝(部分15三体),每个细胞中缺失的染色体片段(部分单体15)和称为环15号染色体的环状结构。