先天性挛缩细长指
先天性挛缩细长指(Congenitalcontracturalarachnodactyly,CCA,OMIM:121050),常染色体显性遗传病,此症与马凡氏综合征(Marfansyndrome,MFS,OMIM:154700)拥有相同的骨骼特征,如多发性关节的先天挛缩、瘦高、细长的手指与脚趾、脊柱侧弯与脊柱后凸、皱耳、漏斗状胸或胸部隆凸,但在关于眼睛与心血管方面的并发症上两者有区别。
先天性挛缩细长指(Congenitalcontracturalarachnodactyly,CCA,OMIM:121050),常染色体显性遗传病,此症与马凡氏综合征(Marfansyndrome,MFS,OMIM:154700)拥有相同的骨骼特征,如多发性关节的先天挛缩、瘦高、细长的手指与脚趾、脊柱侧弯与脊柱后凸、皱耳、漏斗状胸或胸部隆凸,但在关于眼睛与心血管方面的并发症上两者有区别。
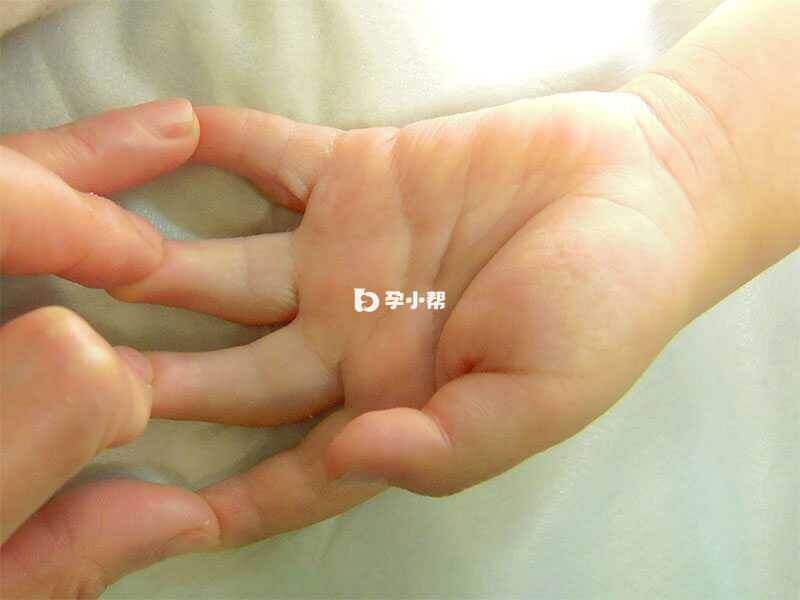
先天性挛缩性细长指是影响身体许多部位的疾病,患有这种病症的人通常具有长肢体(肢体细长症)和细长的手指和脚趾(蜘蛛足样指),他们经常有永久弯曲的关节(挛缩),可以限制他们的臀部,膝盖,脚踝或肘部的运动。
先天性挛缩细长指形成原因
关节的发育是怀孕2从几个月开始,子宫运动减少(子宫发育不全、多、羊水过少)会导致关节挛缩,先天性多关节挛缩[1]是由神经源、肌病和相关组织疾病引起的,先天性肌病、前角细胞疾病、母亲肌无力被认为是肌肉发育不全的原因,先天性多关节挛缩不是一种遗传性疾病,而是一种遗传性疾病(如18三体、脊柱隐裂)会增加关节挛缩的发生率。
先天性挛缩细长指基因介绍
有许多研究表明,CCA主要是由位于第5对染色体上的FBN2基因突变造成的,有大概48种突变被报道,包括31种错义突变和无义突变。这些突变大多位于FBN2基因中部区域外显子22到36之间,若父母双方其中一人为此症患者,不分男女,每一胎皆有1/2的几率会罹患此症。
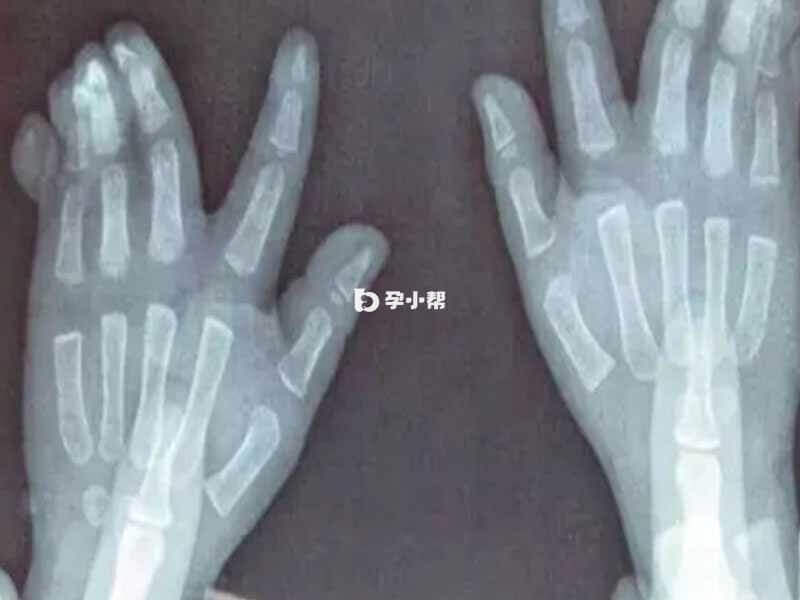
FBN2基因是唯一发现的只与CCA相关的基因,FBN2基因>28kb,含65个外显子,编码2912个氨基酸,由5个不同的结构域组成一个multidomainprotein。这个最大的结构域包含41个钙结合类表皮生长因子基序(Calciumbindingepidermalgrowthfactor-like,cbEGF),所有在FBN2基因上的突变集中在一个特定的区域。
先天性挛缩细长指三代试管
该疾病是一种遗传性疾病,所以最好的就是做三代试管进行阻断遗传,一个案例就是父亲为CCA患者,基因杂合度较低,母亲正常,先证者正常,申请PGD助孕,检测父母双亲、先证者1个、胚胎4个(胚胎4、胚胎5、胚胎10、胚胎13)。最后做的三代试管很成功。
检测方法
一般该疾病的检测方法就是三代试管婴儿S-PGD——三重防护,JBRH解决方案检测特点就是检出率高,适用范围广,检测结果准确,能够有效检测到重组,可无先证者,靶向捕获设计,方案灵活,便于临床成本控制。